The Consequences of Off-Target Binding Are Real. So Are the Solutions.
In this article:
- Can off-target binding cause adverse events?
- What does MPA screening of clinical-stage MAbs reveal?
- Can secreted protein off-targets cause adverse events?
- Do off-targets always cause adverse events?
This article is part 6 of a series about specificity testing.
Summary
Off-target binding doesn’t always cause harm—but when it goes undetected, the consequences can be serious. Published case studies document off-target interactions that caused adverse events in clinical trials, halted drug development programs, and may explain the known safety profiles of approved therapeutics. These cases also demonstrate that when off-targets are identified early, solutions exist: engineering fixes, informed go/no-go decisions, and cleaner IND filings.
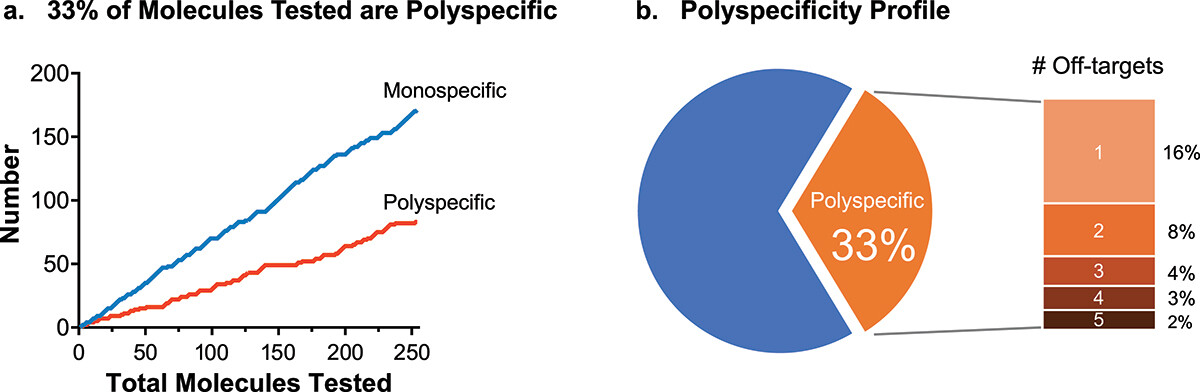
Can off-target binding cause adverse events?
In the previous article, we looked at how common off-target binding is among biotherapeutic drug candidates. Here, we look at what undetected off-target binding can mean in practice.
One of the most well-documented examples of off-target-driven toxicity involves camrelizumab, an anti-PD1 monoclonal antibody developed for cancer immunotherapy. During Phase 1 clinical trials, a majority of treated patients developed severe capillary hemangioma. These abnormal blood vessel growths had no obvious connection to PD1, the antibody’s intended target (Mo et al., 2018).
Retrospective screening of camrelizumab using a cell-based protein array identified the source of the problem: the antibody was also binding VEGFR2, a receptor with a central role in blood vessel formation (Finlay et al., 2019). And the binding wasn’t passive. Camrelizumab acted as an agonist, actively stimulating VEGFR2 to promote vascular neogenesis, which, in vivo, leads to hemangioma.
The causal link was confirmed when patients treated with a VEGFR2 antagonist saw the hemangioma effects resolved (Li et al., 2019). Equally significant: CDR mutagenesis abolished binding to VEGFR2 while preserving high affinity for PD1 (Finlay et al., 2019). Had it been found earlier, the off-target binding could have been engineered away prior to trials in patients.

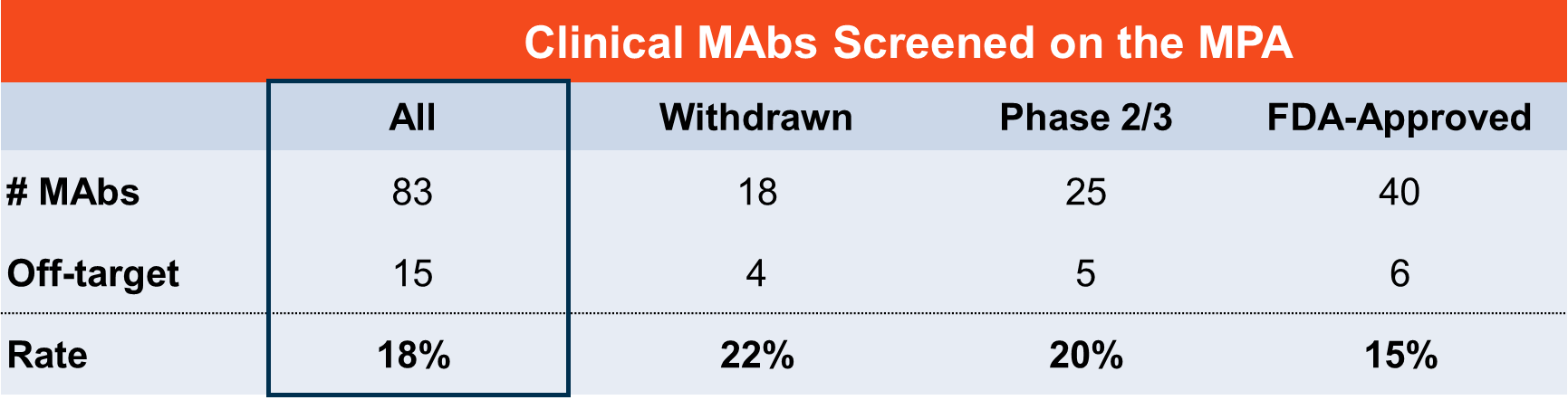
What does MPA screening of clinical-stage MAbs reveal?
Looking more broadly, Membrane Proteome Array (MPA) screening of clinical-stage monoclonal antibodies has identified several cases where off-target binding may explain known adverse events (Norden et al., 2024). Among them:
- A clinical-stage MAb armed with a toxic payload was found to bind an off-target protein with widespread tissue expression—a combination predicted to cause significant toxicity.
- A MAb that was withdrawn from clinical trials due to severe patient adverse events was found to bind an unrelated membrane protein even more strongly than its intended target.
In these cases, the relationship between the off-target interaction and the observed clinical outcome is not fully established. What is clear is that the off-target binding existed, went undetected by prior specificity assessments, and was later identified by MPA screening.
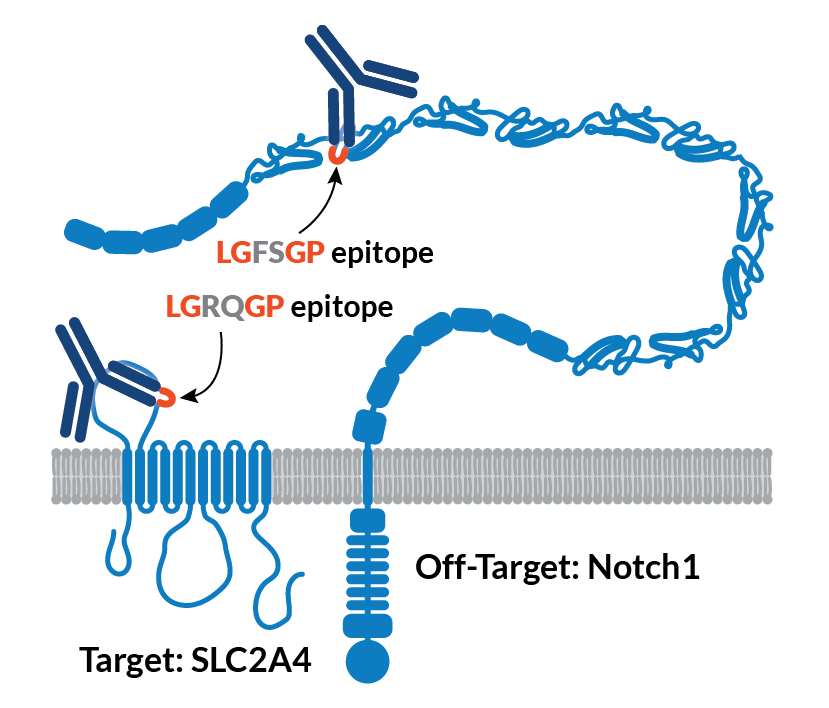
Can secreted protein off-targets pose safety and efficacy risks?
Membrane proteins are the primary focus for specificity testing because off-target binding to them carries the highest safety risk, but off-target binding to secreted proteins can also carry consequences. While off-target binding to secreted proteins is less likely to cause cell death or direct disruption of cell biology, toxicity has still been reported.
In one case, development of ABT-736, an anti-beta-amyloid antibody, was discontinued after it caused severe toxicity in cynomolgus monkeys. This toxicity was later traced to off-target binding to platelet factor 4, a secreted plasma protein (Loberg et al., 2021).
Beyond direct toxicity, off-target binding to abundant circulating proteins can also affect pharmacokinetics and dosing (Bumbaca et al., 2011).
To identify potential secreted off-targets, and to provide a comprehensive picture of your molecule’s interactions with the human body, Integral Molecular offers screening against over 1,200 soluble proteins on the Secreted Proteome Library (SPL). We’ll share more about the SPL in a future article.
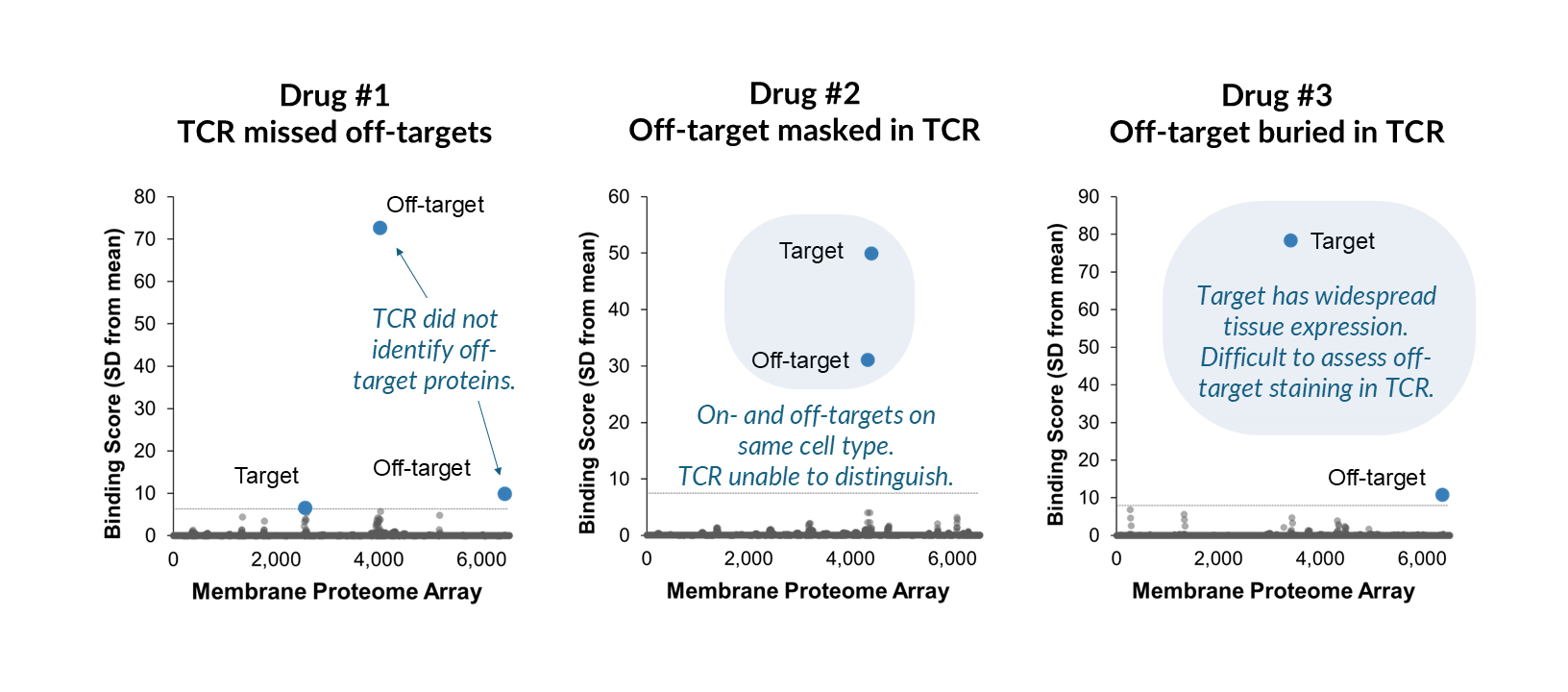
Do off-targets always cause adverse events?
The camrelizumab story could have gone very differently if the VEGFR2 interaction had been identified preclinically. CDR mutagenesis, the same approach that was used retrospectively, could have been applied before patients were enrolled.
This is what earlier, more comprehensive specificity testing enables: not just detection of off-targets, but the opportunity to act on that information while options still exist. For candidates without off-target interactions, MPA results can be included directly in IND filings. For candidates with off-target interactions, the MPA identifies the specific protein involved, enabling a focused investigation rather than a search for an unknown cause. While off-target binding does not always lead to adverse events, it does always warrant investigation.
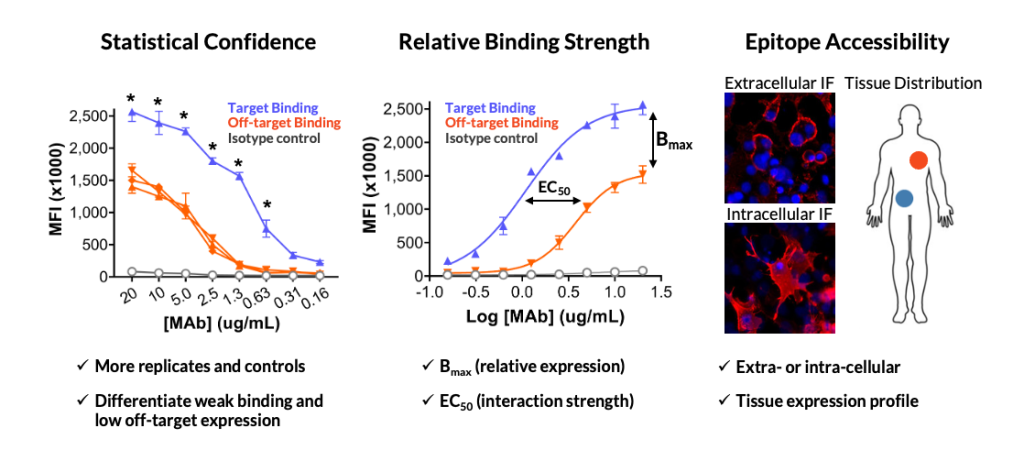
Any investigation into an off-target should consider factors including the relative strength of the off-target interaction compared to on-target binding, the epitope location and accessibility of the off-target protein, and the therapeutic mechanism of action. Integral Molecular’s Enhanced Binding Analysis service provides the quantitative data needed to begin that evaluation.
Understanding these factors will help you understand the associated risk and arm you with the information you need to make decisions. For example, a low-affinity interaction with an intracellular epitope presents a very different risk profile than strong binding to a widely expressed cell-surface protein—particularly for cell-killing modalities like ADCs or CAR-T therapies.
Looking ahead
In the next article, we’ll cover how the MPA library was designed, and what it means for a specificity testing tool to be truly comprehensive.